Predicting Drug Interactions in Pharma With ChemicalX Integration
One of many aspects of the pharmaceutical industry includes researching and developing novel drugs for treating different diseases. This process is often expensive and time-consuming. Companies spend around 25% of their yearly budget on research, but only a small number of experiments turns out to be the next big success.
AstraZeneca is one of the industry leaders in the field of drug discovery and development. You might wonder, how does that relate to Memgraph? The core technology used by the industry leaders in the drug discovery field is in fact graph technology. Graphs are an elegant way to represent drug interactions and components participating there.
Now, let’s introduce a concrete example! ChemicalX is a graph machine learning library based on an in-depth understanding of drug interactions. It provides well-known neural network architectures and datasets for drug pair scoring tasks. This blog post will introduce you to the drug discovery world by answering:
- how drug interactions are researched
- how to successfully develop integration between a graph learning library and Memgraph
- how to run predictive models
Drug interactions and ChemicalX
As any consumer will probably testify, each drug comes with side effects, but the equation/issue becomes a bit more complicated when drugs are combined as they also interact with each other which can additionally unease the consumer. To relieve some of those concerns, each drug comes with instructions and warning lists larger than average blog posts.
Pharmaceutical companies certainly want to enhance the user experience and therefore need to answer some crucial questions before bringing new drug molecules into medical practice:
- Polypharmacy side effects: When combined with different drugs, what side effects can someone expect?
- Drug-drug interactions: What are the effects of drug combinations, and how do they affect the patient?
- Drug synergy: If combined, can the combination of the two drugs create a synergy effect?
Drug trials take years of research by reason of the above points (ChemicalX: A Deep Learning Library for Drug Pair Scoring: Benedek Rozemberczki et.al) and the rigors of regulatory compliance, and their cost has been on a steady rise since the 50s, emburdening pharma, healthcare providers and patients. Nobody wants that. That’s why we use graphs.
Graphs in drug interactions
The idea of modeling drug interactions as graphs can feel highly natural. Different drugs interact with different entities such as proteins, genes, or pathogens, and graph relationships store the information about the interactions.
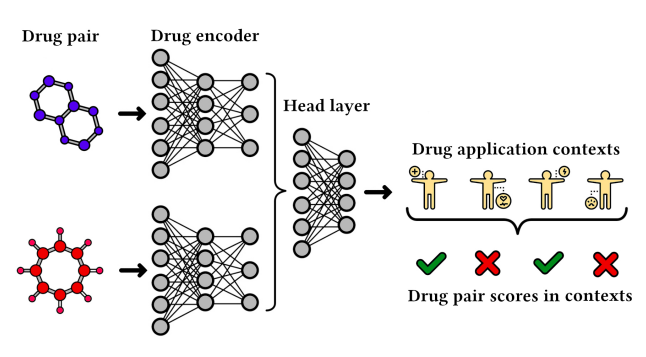 A visualization of scoring the interaction in a drug pair. Relationships between drugs model their combined behavior. (ChemicalX: A Deep Learning Library for Drug Pair Scoring: Benedek Rozemberczki et.al)
A visualization of scoring the interaction in a drug pair. Relationships between drugs model their combined behavior. (ChemicalX: A Deep Learning Library for Drug Pair Scoring: Benedek Rozemberczki et.al)
In the ChemicalX example, datasets focus on modeling the interaction between drugs and are already enriched with additional information. Drugs are described with 4 different pieces of information: a) molecular features, such as charge measures, b) molecular graphs with edges and bonds, c) node features such as type of atoms, and d) edge features describing bonds. Depending on the task, additional context features may also be included, e.g. labeled interactions in terms of drug-drug synergy.
For a task, one can try to predict novel interactions based on the labeled interactions and explore results before conducting expensive real-life experiments. That’s exactly what you’ll see in the next chapter!
Training framework in Memgraph
Let's put drug relationships to the test! Our goal is to build the synergy prediction module with ChemicalX on top of Memgraph. More specifically, the goal is to predict whether the unlabeled combination of drugs will have the synergistic effect.
Prerequisites
It’s recommended to introduce yourself to the following before jumping into development:
- Memgraph Platform - the bundle of Memgraph ecosystem products:
- Memgraph DB - in-memory database
- Memgraph MAGE - a collection of open-source graph algorithms and useful utilities
- Memgraph Lab - integrated graph environment (IGE) that will help us build the prediction module.
Try to keep up with the latest version for best support. However, if something is not working, revert back to version 2.3.1 used in this demo.
- Getting a notion of the query modules
- Downloading the DrugCombDB dataset created for the drug-drug synergy prediction task
Starting the database
We’ll be writing commands necessary to run the Docker instance with a running Memgraph inside. Firstly memgraph/memgraph-platform, an image accompanied by MAGE - a set of graph analytics modules ready to run with openCypher, and Lab - an integrated graph environment. To start a database, simply run the following:
docker pull memgraph/memgraph-platform:2.3.1
docker run -it -p 7687:7687 -p 3000:3000 --name memgraph memgraph/memgraph-platform
Output of these commands should suggest that Memgraph is currently running!
Memgraph Lab is running at localhost:3000
mgconsole 1.1
Connected to 'memgraph://127.0.0.1:7687'
Type :help for shell usage
Quit the shell by typing Ctrl-D(eof) or :quit
memgraph>
Since our intention is to use ChemicalX, we need to install it on our system,in this case it’s Docker. Follow these steps to install the dependencies and enable the ChemicalX library:
docker exec -it -u root mage apt update
docker exec -it -u root mage apt install python3-pip
docker exec -it -u root mage apt install libsm6 libxext6 libxrender-dev
docker exec -it -u root mage pip install torch==1.10.0 torch-scatter -f https://pytorch-geometric.com/whl/torch-1.10.0+cpu.html torchdrug chemicalx
docker exec -it -u root mage chown -R memgraph:memgraph /usr/lib/memgraph/query_modules
If all you are seeing are success messages, let’s move to the data fetching part!
Getting the data
Memgraph Lab interface enables users to easily run different queries and offers a large number of possibilities related to the interaction with the Memgraph ecosystem.
The data consists of chemical compounds and their descriptions. The edges play a key role in describing the interaction between the compounds with context information and the label of interest (in this case, the synergy effect). If you want to learn more about different compounds, we suggest exploring the PubChem database by providing the node ID as a reference. Nodes are populated with common chemical names and their SMILES identifier. As said before, molecular structure features are pre-computed by ChemicalX developers and are stored as vector in the features property.
Data ingestion should be fairly straightforward and quite fast. The dataset is hosted by Memgraph in cypherl format, and therefore can be downloaded with a few clicks in Memgraph Lab by exploring the Datasets section in the left side menu. The dataset in Lab which will be used is called “Drug combinations database”, and when you hover over it and click on the Load button, the dataset will be imported into Memgraph.
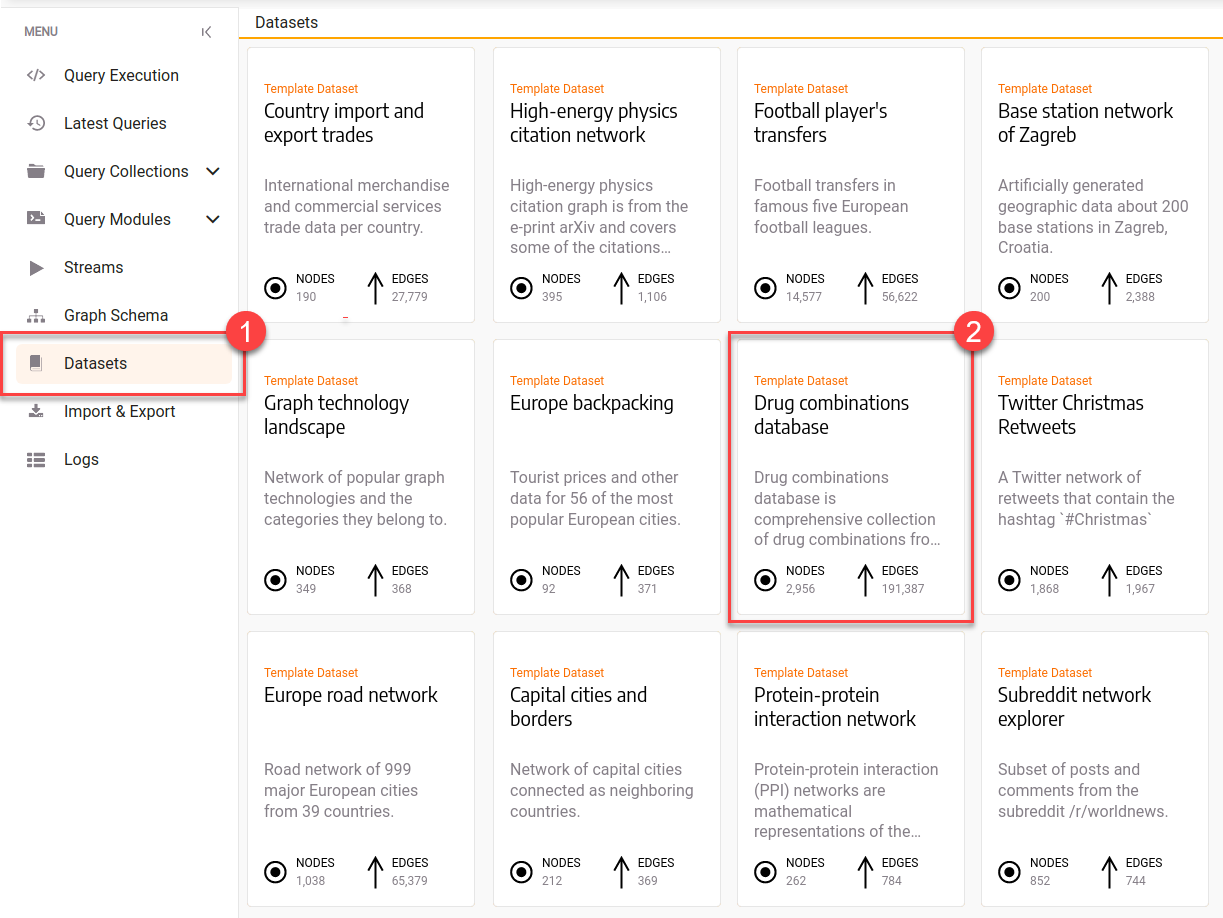
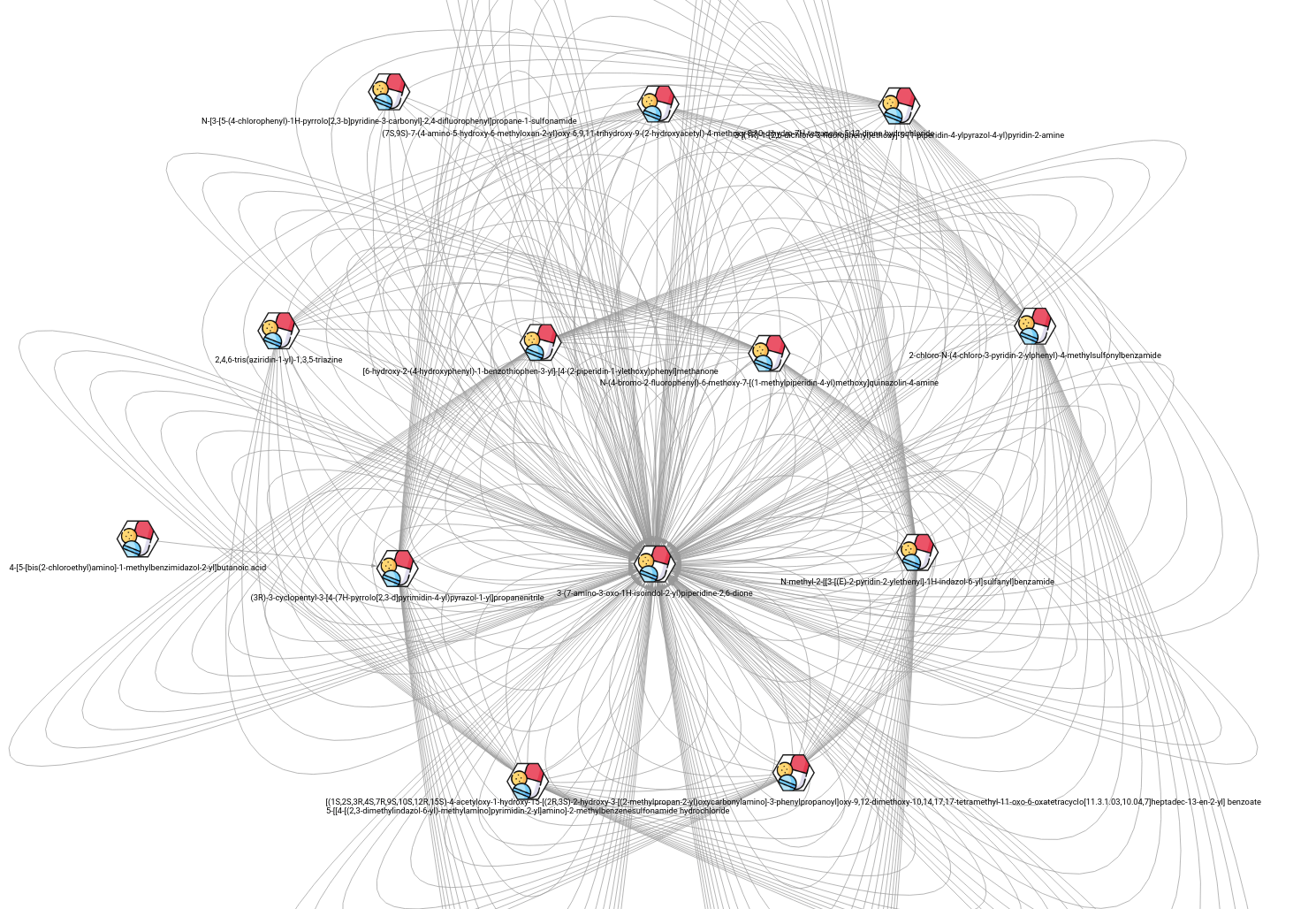 Illustration of drug interaction network. Drugs are hexagon nodes and interaction between them denote synergy effect within some context.
Illustration of drug interaction network. Drugs are hexagon nodes and interaction between them denote synergy effect within some context.
Writing the module
Now for the most interesting part of this blog - creating a module! With the provided Memgraph API you can easily communicate with the storage, and access the entities stored in the DB. This way Memgraph offers access to the C++, Rust, and Python for communication between the DB and the user.
Since Memgraph Lab is a powerful IGE (integrated graph environment), it enables developing modules within it. Firstly, let’s create a file called memchemicalx by pressing the + New Module button. The module will then sync with Memgraph’s storage and the file will be updated once the Save button is pressed.
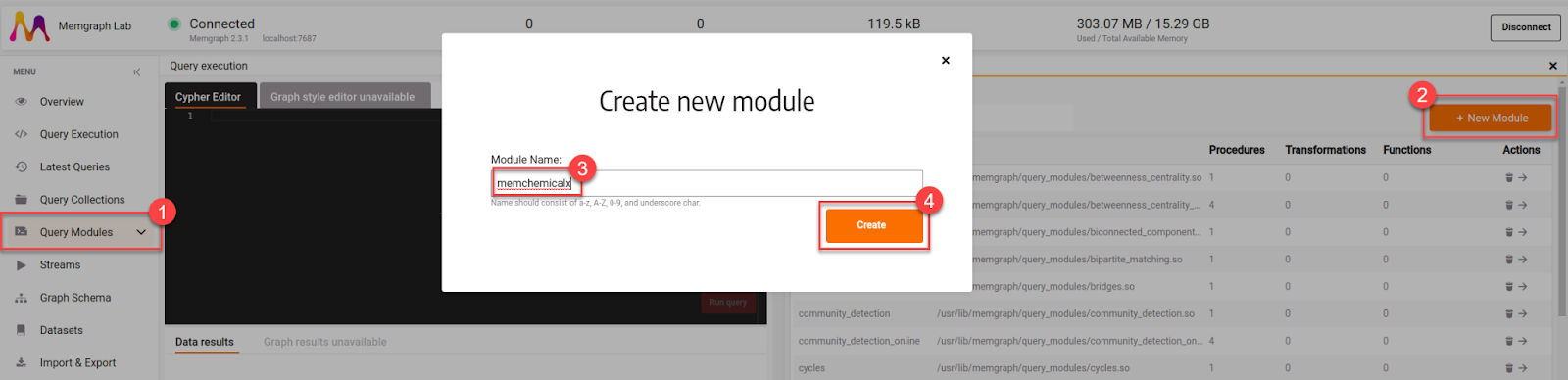
At first glance, we are able to see the signature of the default so-called read procedure - custom method created for using the graph in read-only mode, meaning that writing the data is prohibited. This behavior is denoted by annotation @mgp.read_proc.
We’ll be writing a module capable of training and predicting synergy interactions between drugs. Let’s start by writing just the signatures of needed read-procedures. Signature should contain the definition of input and output parameters types. First argument is the graph context, always present in Memgraph API, followed by custom parameters needed to create an ML method.
- Training procedure signature:
@mgp.read_proc
def train(
ctx: mgp.ProcCtx,
learning_rate: float = 1e-3,
batch_size: int = 64,
epochs: int = 100,
) -> mgp.Record(loss_value=mgp.Number):
- Prediction procedure signature:
@mgp.read_proc
def predict(
ctx: mgp.ProcCtx,
edges: mgp.List[mgp.Edge] = None,
batch_size: int = 32,
) -> mgp.Record(predictions=list):
This is the base for our prediction module. We intend to first call the training method to initialize training data and model, and then when the model is trained on data, predict on newly added data points.
Like in any other machine learning framework, the crucial part is implementing the data generator. Since the dataset is pulled from Memgraph, the implementation of the data generator will be a little different. In the initialization phase, we extract the graph vertices information by iterating the nodes by calling ctx.graph.vertices. All nodes should have the similar format specified for ChemicalX described in the introduction. To extract interactions, labeled triples are pulled from edges currently being stored in the system.
import mgp
from typing import Union
from collections import defaultdict
import torch
import numpy as np
from sklearn.preprocessing import LabelBinarizer
from chemicalx.models import DeepSynergy, Model
from chemicalx.data import (
BatchGenerator,
DatasetLoader,
DrugFeatureSet,
ContextFeatureSet,
LabeledTriples,
)
class MemgraphDatasetLoader(DatasetLoader):
DRUG_1 = "drug_1"
DRUG_2 = "drug_2"
ID = "id"
SMILES = "smiles"
FEATURES = "features"
CONTEXT = "context"
LABEL = "label"
def __init__(self, ctx: mgp.ProcCtx, edges: mgp.List[mgp.Edge] = None):
"""Instantiate the Memgraph dataset loader specific for drug-drug interaction."""
self.drug_data = {}
self.interaction_data = defaultdict(list)
# Process drugs
for vertex in ctx.graph.vertices:
self.drug_data[str(vertex.properties.get(MemgraphDatasetLoader.ID))] = {
MemgraphDatasetLoader.SMILES: vertex.properties.get(
MemgraphDatasetLoader.SMILES
),
MemgraphDatasetLoader.FEATURES: np.array(
vertex.properties.get(MemgraphDatasetLoader.FEATURES)
).reshape(1, -1),
}
# Process drug interactions
for edge in self.edge_generator(edges if edges else ctx):
context = edge.properties.get(MemgraphDatasetLoader.CONTEXT)
label = edge.properties.get(MemgraphDatasetLoader.LABEL)
from_vertex = edge.from_vertex
to_vertex = edge.to_vertex
self.interaction_data[MemgraphDatasetLoader.DRUG_1].append(
str(from_vertex.properties.get(self.ID))
)
self.interaction_data[MemgraphDatasetLoader.DRUG_2].append(
str(to_vertex.properties.get(self.ID))
)
self.interaction_data[MemgraphDatasetLoader.CONTEXT].append(context)
self.interaction_data[MemgraphDatasetLoader.LABEL].append(label)
@staticmethod
def edge_generator(source: Union[mgp.ProcCtx, mgp.List[mgp.Edge]]):
if type(source) == mgp.ProcCtx:
for vertex in source.graph.vertices:
for edge in vertex.out_edges:
yield edge
else:
for edge in source:
yield edge
def get_drug_features(self) -> DrugFeatureSet:
"""Get the drug feature set."""
return DrugFeatureSet.from_dict(self.drug_data)
def get_labeled_triples(self) -> LabeledTriples:
"""Get the labeled triples dataframe. The labels correspond to synergy effect."""
return LabeledTriples(self.interaction_data)
@staticmethod
def get_context_features(interaction_data) -> ContextFeatureSet:
"""Get the context feature set out of interaction data."""
context = list(set(interaction_data[MemgraphDatasetLoader.CONTEXT]))
context_data = {
label: encoding
for label, encoding in zip(context, LabelBinarizer().fit_transform(context))
}
return ContextFeatureSet.from_dict(context_data)
MemgraphDatasetLoader is fully customized to work with the data created from the DrugCombDB dataset. The previously explained drug and context features are accessible from Loader as well as labeled triplets containing the information about the domain-specific labels.
Our plan is to use the full available information stored in Memgraph as the train set for the ML model. That’s why we intentionally left out some labeled data in order to have examples to test our model’s performance in predicting drug interactions. Our training interface should look something like this:
CALL memchemicalx.train(1e-4, 1024, 100) YIELD loss_value;
Where the arguments are the training edges, the learning rate, the batch size, and the number of training epochs. To match this query, let’s implement the core functionality - model training. To ensure persistence, declare the model as a global variable in the module. Implementation follows the typical example of machine learning training: the generator generates the batches of data, followed by the prediction step and gradient backpropagation used to update the model’s parameters. The training uses simplistic binary cross-entropy loss function and adam optimizer, while the model used is called DeepSynergy (Preuer, K., et al. (2018). DeepSynergy: predicting anti-cancer drug synergy with Deep Learning), created for assessing the drug synergy effect in cancer treatment.
model: Model = None
context_set: ContextFeatureSet = None
@mgp.read_proc
def train(
ctx: mgp.ProcCtx,
learning_rate: float = 1e-3,
batch_size: int = 64,
epochs: int = 100,
) -> mgp.Record(loss_value=mgp.Number):
global model, context_set
# Initialize loader
print("Dataset loading...")
loader = MemgraphDatasetLoader(ctx)
drug_set = loader.get_drug_features()
interaction_set = loader.get_labeled_triples()
context_set = MemgraphDatasetLoader.get_context_features(loader.interaction_data)
generator = BatchGenerator(
batch_size=batch_size,
context_features=True,
drug_features=True,
drug_molecules=False,
context_feature_set=context_set,
drug_feature_set=drug_set,
labeled_triples=interaction_set,
)
print("Dataset loaded...")
# Model specific for DrugCombDB dataset
model = DeepSynergy(context_channels=112, drug_channels=256)
model.train()
# Initialize training parameters
loss = torch.nn.BCELoss()
optimizer = torch.optim.Adam(model.parameters(), lr=learning_rate)
loss_avg = 0
print("Model created, start training...")
for epoch in range(epochs):
loss_total, num_batches = 0, 0
for batch in generator:
optimizer.zero_grad()
prediction = model(
batch.context_features,
batch.drug_features_left,
batch.drug_features_right,
)
loss_value = loss(prediction, batch.labels)
loss_value.backward()
optimizer.step()
num_batches += 1
loss_total += float(loss_value)
loss_avg = float(loss_total) / float(num_batches)
print(f"Epoch {epoch + 1}/{epochs}, Loss: {loss_avg}")
return mgp.Record(loss_value=loss_avg)
Now, the module should be sufficient to train our model. As the result, you’ll get the output of the training loss calculated from the training data. But choose wisely! Parameters such as learning rate and the number of epochs can make a difference while training.
Predict the interactions
The goal of this blog was to present how to predict whether two drugs will create a synergic effect on treatment, i.e. if they will help enhance the therapeutic effect. After training our machine learning model in the previous chapter, this chapter will focus on the prediction of synergistic effects.
We removed some of the labeled data on purpose, and now we’ll add it to simulate prediction on new data points, similarly to splitting the dataset into training and test set. But, what we are missing is the prediction procedure.
Predicting the synergy on new data means using the trained model defined above without updating the parameters on call (without training). This is again done by creating a generator that will batch and iterate the data ready for the prediction task. Batch prediction is used because it is faster than running the execution one-by-one data point. After predicting, predictions are stored in a similar order as they were sent as input.
@mgp.read_proc
def predict(
ctx: mgp.ProcCtx,
edges: mgp.List[mgp.Edge],
batch_size: int = 16,
) -> mgp.Record(predictions=mgp.List[mgp.Number]):
global model, context_set
if model is None or context_set is None:
raise RuntimeError("The model is not initialized!")
model.eval()
loader = MemgraphDatasetLoader(ctx, edges)
drug_set = loader.get_drug_features()
triples = loader.get_labeled_triples()
generator = BatchGenerator(
batch_size=batch_size,
context_features=True,
drug_features=True,
drug_molecules=False,
context_feature_set=context_set,
drug_feature_set=drug_set,
labeled_triples=triples,
)
predictions = [0] * len(edges)
for batch in generator:
prediction_batch = model(
batch.context_features, batch.drug_features_left, batch.drug_features_right
)
prediction_batch = prediction_batch.detach().cpu().numpy()
for (index, _), prediction in zip(batch.identifiers.iterrows(), prediction_batch):
predictions[index] = float(prediction)
return mgp.Record(predictions=predictions)
To run a prediction task, first, add data points into the database:
MATCH (n:Drug {id: 68210102})
MATCH (n1:Drug {id: 11598628})
CREATE (n)-[:NEW_INTERACTION {context:'A-673', label:0.0}]->(n1);
MATCH (n:Drug {id: 5281607})
MATCH (n1:Drug {id: 11598628})
CREATE (n)-[:NEW_INTERACTION {context:'A-673', label:0.0}]->(n1);
MATCH (n:Drug {id: 515328})
MATCH (n1:Drug {id: 16654980})
CREATE (n)-[:NEW_INTERACTION {context:'U-HO1', label:1.0}]->(n1);
MATCH (n:Drug {id: 9806229})
MATCH (n1:Drug {id: 148198})
CREATE (n)-[:NEW_INTERACTION {context:'U-HO1', label:1.0}]->(n1);
And use the prediction function to predict the possible synergy effects when two drugs are used together:
MATCH ()-[e:NEW_INTERACTION]->()
WITH COLLECT(e) AS edges
CALL memchemicalx.predict(edges) YIELD edge, prediction
RETURN startNode(edge) AS drug_1,
endNode(edge) AS drug_2,
edge AS interaction,
prediction,
edge.label AS label;
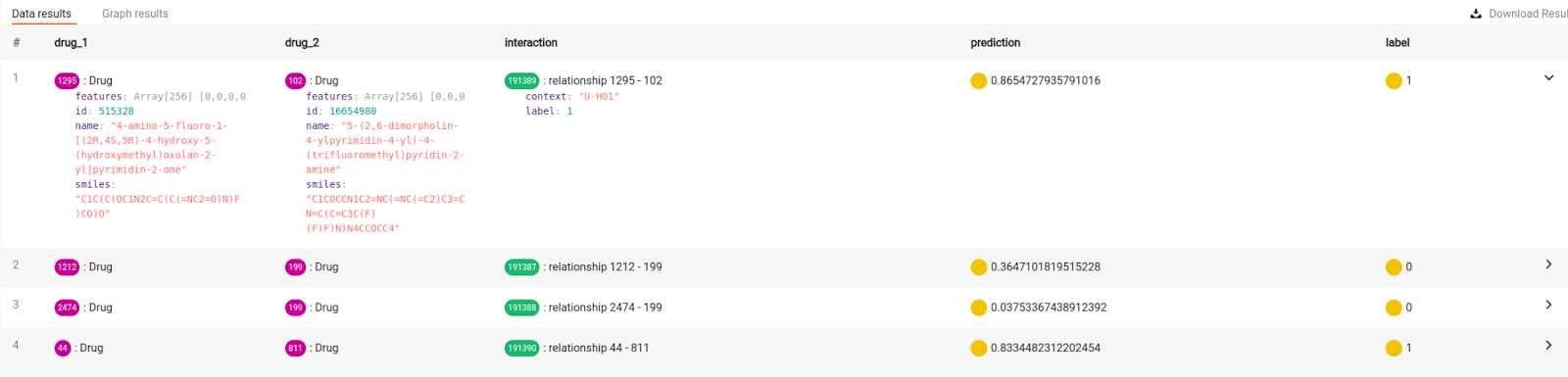
As seen in the photo below, the model is quite successful in assessing the synergy effect on newly added interactions. If the prediction threshold is set at 0.5, the model gets every interaction right!
Conclusion
The pharmaceutical industry is the leader in drug development as well as modern graph research. Products like ChemicalX offer high flexibility for the user even in this early development stage. Future graph research will involve even bigger data with even more complexity, and the evolution of graph databases will play a key role in data storage. We as Memgraph are happy to be highly extensible and offer easy integration with novel tools such as ChemicalX.
If you need a hand with using Memgraph here’s our Discord server or GitHub community. Feel free to engage, ask questions and challenge us to work harder to help you on the path to being the graph expert. Happy coding!